Tag: Protein Design
-

Open Philanthropy awards $11.3 million to the Institute for Protein Design
The funds will support our technological revolution in protein design and enable the development of a universal flu vaccine. The $11.3 gift is one of the largest made to date by the San Francisco-based philanthropy in support of science. It is also the first to go to UW Medicine. The gift comes in two parts:…
-

Unleashing the Power of Synthetic Proteins
Published today in Science Philanthropy Alliance, David Baker, Director of the Institute for Protein Design describes how the opportunities for computational protein design are endless — with new research frontiers and a huge variety of practical applications to be explored, from medicine to energy to technology. This is an exciting time as we are undergoing a technological revolution…
-

Hyper-stable Designed Peptides and the Coming of Age for De Novo Protein Design
Small constrained peptides combine the stability of small molecule drugs with the selectivity and potency of antibody-based therapeutics. However, peptide-based therapeutics have largely remained underexplored due to the limited diversity of naturally occurring peptide scaffolds, and a lack of methods to design them rationally. New computational design and wet lab methods developed at the Institute for…
-
Computational Design of a pH Sensitive Antibody Binder
Purification of antibody IgG from crude serum or culture medium is required for virtually all research, diagnostic, and therapeutic antibody applications. Researchers at the Institute for Protein Design (IPD) have used computational methods to design a new protein (called “Fc-Binder”) that is programed to bind to the constant portion of IgG (aka “Fc” region) at basic…
-
Computational Protein Design To Improve Detoxification Rates Of Nerve Agents
V-type nerve agents are among the most toxic compounds known, and are chemically related to pesticides widespread in the environment. Using an integrated approach, described in an ACS Chemical Biology paper entitled Engineering V-type nerve agents detoxifying enzymes using computationally focused libraries, Dr. Izhack Cherny, Dr. Per Greisen, and collaborators increased the rate of nerve…
-
Follow the Institute for Protein Design on Facebook and Twitter
Visit us on Facebook and follow us on Twitter @UWproteindesign
-
Professor David Baker Provides an In Depth Discussion on Protein Design
Prof. David Baker, Head of the UW Institute for Protein Design, HHMI Investigator provides an in depth discussion on the design of protein structures, functions and assemblies. Click here to watch the video.
-
Institute for Protein Design Infograph
[soliloquy id=”345″]
-
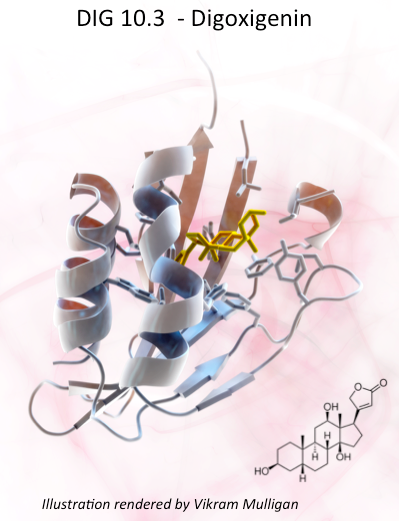
One Small Molecule Binding Protein, One Giant Leap for Protein Design
Reported on-line in Nature (Sept. 4, 2013) researchers at the Institute for Protein Design describe the use of Rosetta computer algorithms to design a protein which binds with high affinity and specificity to a small drug molecule, digoxigenin a dangerous but sometimes life saving cardiac glycoside. Learn more at this link.