Today we report the design of protein sequences that adopt more than one well-folded structure, reminiscent of viral fusion proteins. This research moves us closer to creating artificial protein systems with reliable moving parts.
In nature, many proteins change shape in response to their environment. This plasticity is often linked to biological function. While computational protein design has been used to create molecules that fold to a single stable state and to re-engineer natural proteins to alter their dynamics or fold, the design from scratch of closely related sequences that adopt well-defined but divergent structures has remained an outstanding challenge.

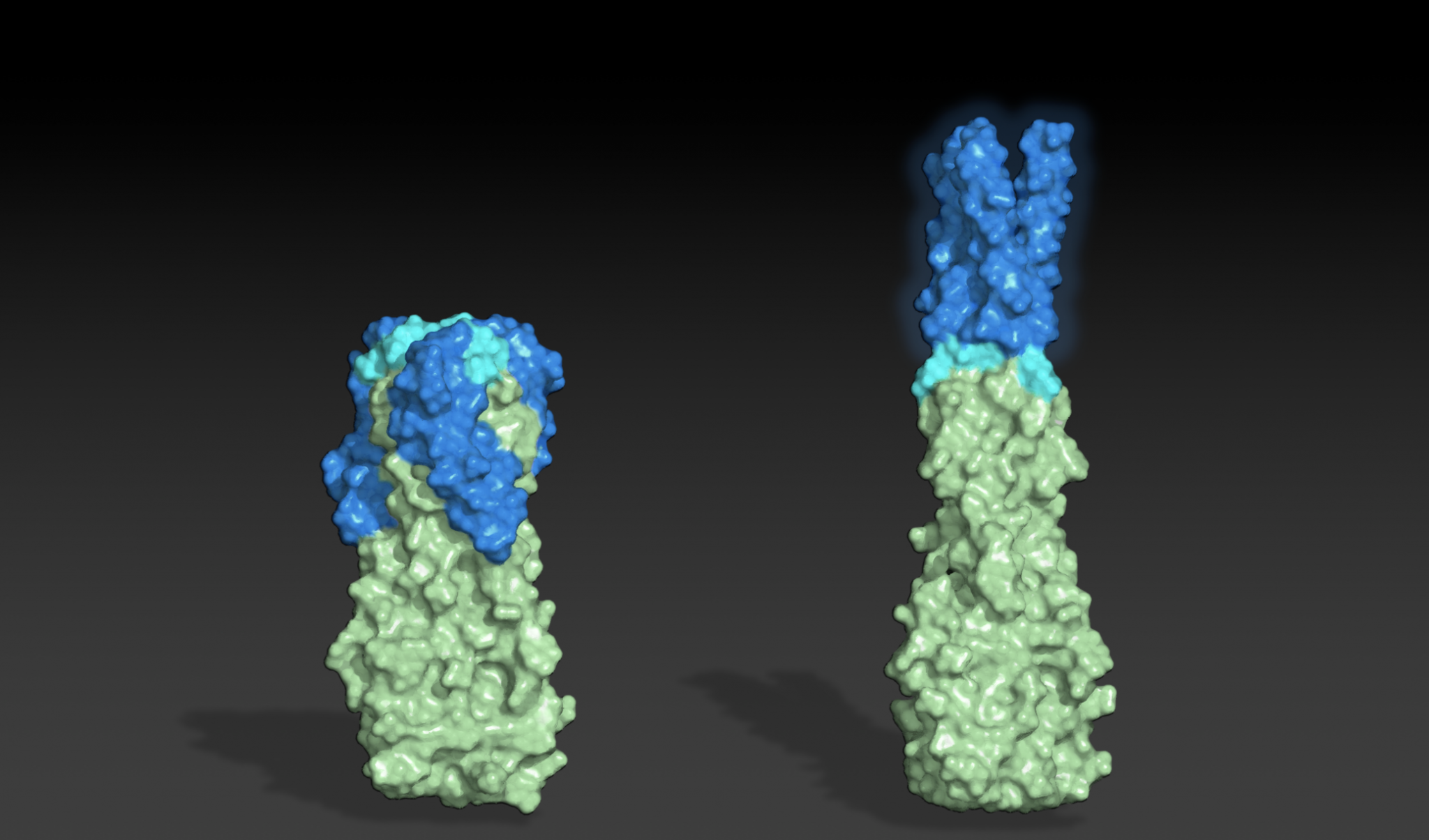
To create shape-shifting proteins, a team led by recent Baker lab postdoc Kathy Wei, Ph.D., began by identifying sets of amino acid sequences predicted to fold into very different structures — in this case, pairs of cylindrical helical bundles with different lengths.
“We knew from the beginning that we wanted a sequence to transform between a short state with helical “arms” pointed “down” and a long state with helical “arms” pointed “up”. The plan was to use established protocols to first design different proteins that are in each of the two states and then mutate the sequences of these two starting points toward each other until we found a sequence that could fold into both states,” said Wei.
After rounds of design on the computer and testing in the lab, the team succeeded in creating a single molecule that could be seen in both states.
“One of the main challenges for this project was finding a way to tell if the proteins took on the shape they were designed to be in. High-throughput screening methods tend to rely on an enzymatic property of a protein. Since these designed proteins only differed in their shapes, we had to use crystallography and NMR to check their folding, which is a slow process and not guaranteed to yield results.”
“While we found a really promising protein sequence that we can measure in both of the designed states, it’s surprisingly much less dynamic than we would’ve expected. Next, we want to understand how to make the conformational changes more dynamic and how to trigger them in a controlled manner.“
The team included scientists from the University of Washington, UC Berkeley, UC Santa Cruz, and Stanford. Their work was supported by the NIH, DOE, HHMI and the Chan Zuckerberg Biohub.
Computational design of closely related proteins that adopt two well-defined but structurally divergent folds. Kathy Y. Wei, Danai Moschidi, Matthew J. Bick, Santrupti Nerli, Andrew C. McShan, Lauren P. Carter, Po-Ssu Huang, Daniel A. Fletcher, Nikolaos G. Sgourakis, Scott E. Boyken, and David Baker. PNAS.
Read the full report: https://www.pnas.org/content/early/2020/03/17/1914808117 PDF