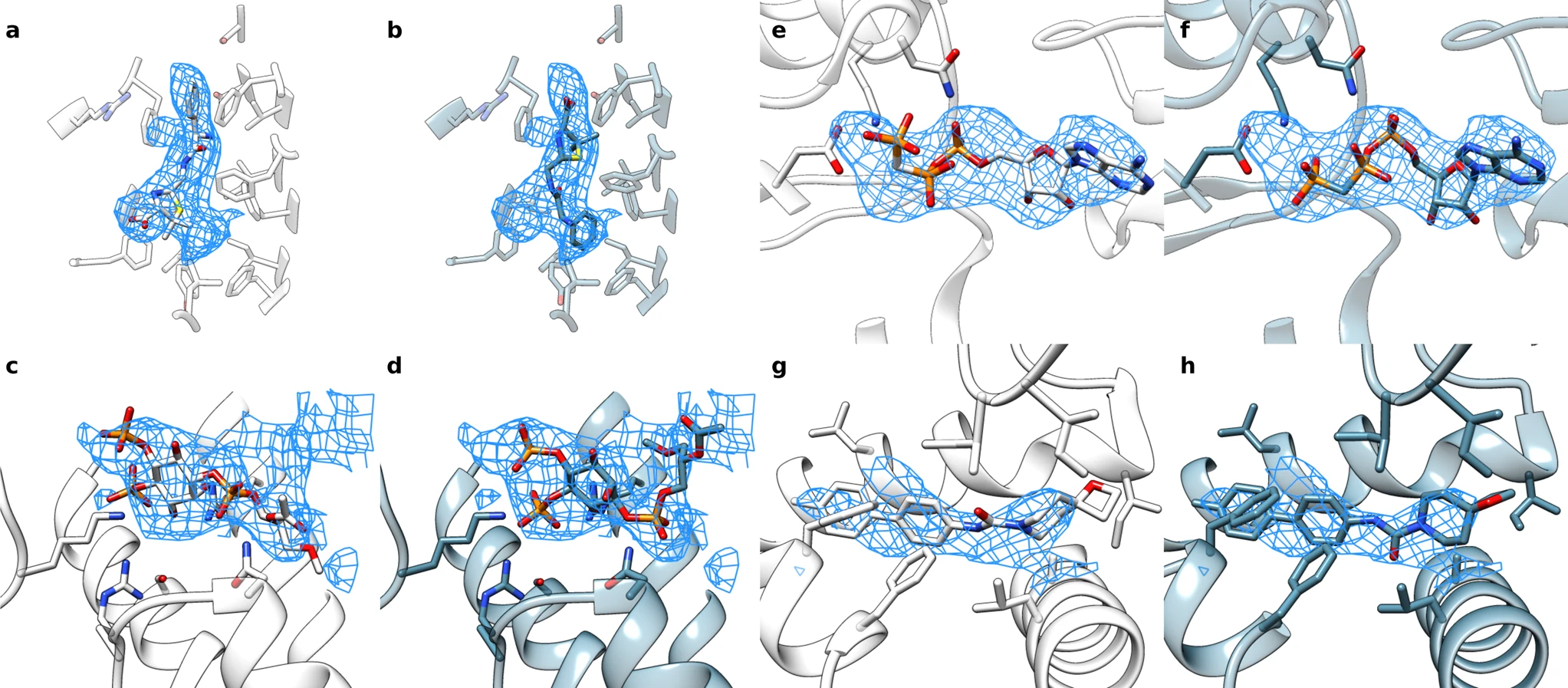
Under ideal conditions, cryo-electron microscopy can be used to determine protein structures at near-atomic resolution. But conditions are not always perfect. To help researchers make use of medium-resolution cryo-electron density maps, scientists in the DiMaio Lab have developed EMERALD, which is a new software tool that can accurately and automatically produce deposition-ready small molecule models into cryoEM maps.
Benchmarked on over 1,000 ligand-bound proteins, EMERALD identified a confident solution in 62% of EMDB entries. In some cases, it identified alternate models that were supported by external data. Led by graduate student Andrew Muenks, this research was published in Nature Communications and included collaborators from the Veesler Lab at UW.

All methods described in the paper are available as part of Rosetta, using weekly releases after February 5, 2023 (version 2023.06 or later). The Rosetta XML files and flags for running all the refinements discussed in this manuscript are included in Methods. A demo for running EMERALD is included in Supplementary Data 2.
Read the full report:
Automatic and accurate ligand structure determination guided by cryo-electron microscopy maps (Open Access)